1. 受试者能够按附录1所述签署知情同意,包括遵守知情同意书和本方案中列出的要求和限制条件
2. 受试者(女性,不考虑绝经状态,以及男性)签署知情同意书时年龄≥18岁
3. 患有多中心性(同一乳房的不同象限内存在两个或多个肿瘤病灶)和/或多灶性(同一乳房的单个象限内存在两个或多个肿瘤病灶)乳腺癌的受试者,如果所有肿瘤检查结果均符合ER阳性和HER2阴性的病理学标准,则有资格参加研究–患有双侧同时性浸润性乳腺癌的受试者,如果所有经组织病理学检查的肿瘤均符合ER阳性和HER2阴性的病理学标准,则有资格参加研究。
4. 根据在当地进行的原发性疾病标本免疫组化(IHC)评估,以及按照美国临床肿瘤学会(ASCO)/美国病理学家协会(CAP)指南(AllisonKHetal2020)的定义,即染色阳性肿瘤细胞≥1%,确认受试者存在ER+肿瘤。
5. 根据在当地进行的原发性疾病标本评估,以及按照ASCO/CAP指南(WolffACetal2018)的定义,确认受试者存在HER2-肿瘤
6. 受试者必须接受过原发性乳腺肿瘤的根治性手术。除下述情况外,经当地病理学家判断,切除标本的边缘在组织学上必须无浸润性肿瘤和/或导管原位癌(DCIS)成分。如果病理学检查发现切除边缘存在肿瘤,可能需要额外进行切除,使边缘无肿瘤。如果再次切除后切缘仍存在肿瘤,则受试者必须接受乳房切除术才有资格参加研究。需要注意的是,边缘为小叶原位癌(LCIS)阳性的受试者有资格参加研究,无需额外切除。–对于接受乳房切除术或局部广泛切除术(深切缘紧贴胸肌筋膜)的受试者,只要在进入研究前接受过胸壁放疗,则显微镜下切缘阳性的受试者有资格参加研究。如果无任何大体病变残留(根据当地指南进行放疗),前切缘阳性的受试者可能有资格参加研究。–对于疑有锁骨上或内乳淋巴结转移的受试者,应根据当地标准指南进行治疗。–如果给予放射治疗(例如,在乳房切除术后或乳房肿瘤切除术后),应根据标准指南进行。
7. 接受或将要接受辅助化疗的受试者必须在随机化前完成辅助化疗。受试者还可能接受过新辅助化疗。末次辅助化疗给药至随机化之间需要至少21天的洗脱期。–允许不适合辅助化疗或拒绝化疗的受试者入组。
8. 既往抗癌治疗或外科手术的所有急性毒性反应均消退至NCICTCAEv5.01级或更好水平的受试者(脱发、≤2级周围神经病变、关节痛或研究者认为不会对受试者造成安全性风险的其他毒性除外)
9. 接受过(新)辅助化疗和/或接受过手术且既往未接受过ET的受试者有资格参加研究,但应在确定性乳腺癌手术后12个月内入组。
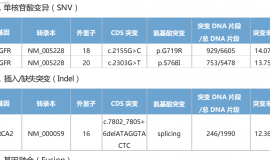
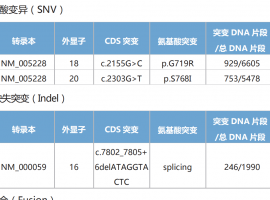
10. 确认受试者有适合生物标志物检测的肿瘤组织标本(即代表性的存档福尔马林固定、石蜡包埋[FFPE]组织蜡块[首选]或含未染色、新鲜连续切片的15-20张玻片),同时需要提供去识别化病理报告。在接受全身性治疗前获得的乳腺肿瘤组织样本必须在筛选期间交由中心病理学实验室进行检测(有关肿瘤组织要求的详细信息,参见第8.7节),以根据Ki67评分确定受试者的合格性。
11. 无病理性受累淋巴结(pN0)的受试者的原发性肿瘤必须大于1cm,且必须满足表5(中危)中概述的至少一个特征–患有病理性淋巴结阳性疾病(镜下和/或肉眼可见肿瘤侵犯)的受试者有资格参加研究,并将根据表5中定义的标准按中危或高危分层。将同侧内乳和/或锁骨上淋巴结阳性的受试者归为高危,不考虑腋窝淋巴结受累情况。对于同时接受(新)辅助治疗的受试者,在初次诊断时允许出现细胞学肿瘤侵犯
12. 按照表5(高危)所示,存在T4原发性肿瘤和任何淋巴结的受试者
13. 美国东部肿瘤协作组(ECOG)体能状态评分为0、1或2的受试者
14. 能够且愿意吞咽口服药物且可将药物保留在体内的受试者
15. 按照以下标准定义,器官功能恰当的受试者:–ANC≥1×109/L(1000/μL);–血小板计数≥100×109/L(≥100,000/μL);–AST和血清ALT≤2×ULN;–ALP≤2倍ULN;–血红蛋白≥80g/L(≥8g/dL);–血清胆红素≤1.5×ULN,以下情况除外:受试者已知患有Gilbert综合征:≤3×ULN–按机构指南计算,肌酐清除率估计值≥60mL/min–INR<1.5×ULN且PTT(或aPTT)<1.5×ULN对于需要接受华法林抗凝治疗的受试者,INR需稳定在2至3之间。对于接受肝素治疗的受试者,PTT(或aPTT)需在1.5×至2.5×ULN之间(或受试者在开始肝素治疗前的数值)。如果人工心脏瓣膜需要抗凝治疗,则INR需稳定在2.5至3.5之间。
16. 对于有生育能力的女性受试者:同意保持禁欲(禁止异性性交)或采取避孕措施(分配至他莫昔芬组的女性受试者也必须同意不捐献卵子)的受试者,其定义如下:女性受试者必须在治疗期间和giredestrant末次给药后9天内或当地处方指南规定的TPC末次给药后时间段内(即他莫昔芬末次给药后60天;来曲唑或阿那曲唑末次给药后21天;依西美坦末次给药后30天)保持禁欲或使用年失败率<1%的非激素类避孕方法,且女性受试者须在他莫昔芬末次给药后60天内不捐献卵子。如果女性受试者处于绝经后期、尚未达到完全绝经状态(连续≥12个月无月经,且除绝经之外无其他原因),且未接受过永久绝育手术(即摘除卵巢、输卵管、和/或子宫)或研究者确定的其他原因(如米勒管发育不全),则认为该女性受试者具有生育能力。可对具有生育能力的定义进行调整,以便与当地指南或法规保持一致。年失败率<1%的非激素类避孕方法包括双侧输卵管结扎、男性绝育术和含铜宫内节育器。应根据临床试验持续时间、个人的首选和惯常生活方式评价禁欲的可靠性。周期性禁欲(如日历法、排卵期法、症状体温法或排卵后安全期法)和体外射精均不是适当的避孕方法。LHRH激动剂不是适当的避孕方法。如果当地指南或法规有要求,应在当地知情同意书中提供当地认可的适当避孕方法和有关禁欲可靠性的信息。
17. 对于入组的男性受试者:同意保持禁欲(禁止异性性交)或使用避孕套,并同意不捐精的受试者,其定义如下:如果女性伴侣具有生育能力或已怀孕,男性受试者必须在治疗期间和giredestrant末次给药后9天内或按当地处方指南规定的TPC末次给药后时间段内(即他莫昔芬末次给药后90天;来曲唑或阿那曲唑末次给药后21天;依西美坦末次给药后30天)保持禁欲或使用避孕套,以避免胚胎暴露药物。男性受试者在此期间不得捐献精子。应根据临床试验持续时间、个人的首选和惯常生活方式评价禁欲的可靠性。周期性禁欲(如日历法、排卵期法、症状体温法或排卵后安全期法)和体外射精均不是预防药物暴露的适当方法。如果当地指南或法规有要求,应在当地知情同意书中提供有关禁欲可靠性的信息。
18. 受试者愿意并能够使用ePRO电子设备或应用程序
19. 对于在中国扩展招募期入组的受试者:具有中国血统、现居住在中国大陆、中国香港或中国台湾的居民。
排除标准
1. 妊娠期或哺乳期受试者,或计划在研究期间或giredestrant末次给药后9天内,或按当地处方指南规定的TPC末次给药后时间段内怀孕的受试者。在开始研究治疗前7天内,具有生育能力的女性受试者的血清妊娠试验结果必须呈阴性。
2. 受试者在开始研究治疗前28天内接受过试验性治疗,或现已入组申办方认为在科学或医学上与本研究不相容的任何其他类型的医学研究。
3. 受试者接受或计划接受CDK4/6i作为辅助治疗。
4. 受试者患有活动性心脏病或具有心功能障碍病史,包括以下任何一项:–具有症状性心动过缓病史(筛选前5年内)或症状或筛选时静息心率<50bpm;–如果静息心率≥50bpm,则可允许受试者接受稳定剂量的β受体阻滞剂或钙通道拮抗剂治疗既存基线疾病(例如,高血压);–随机化前12个月内有心绞痛、症状性心包炎、心肌梗死或任何心律失常(例如室性、室上性、结性心律失常或传导异常)病史;–有确诊的充血性心力衰竭(纽约心脏学会II-IV级)或心肌病史;–三次ECG检查的平均值显示按Fridericia公式校正后QT间期>470ms,有长或短QT综合征、Brugada综合征病史,或已知有校正后的QT间期延长或尖端扭转型室性心动过速病史;–存在研究者认为具有临床意义的ECG异常史或ECG异常,包括完全左束支传导阻滞、二度或三度心脏传导阻滞、病窦综合征或既往心肌梗死的证据。
5. 患有一度心脏传导阻滞的受试者,在咨询心脏病专家并确定不存在额外的心脏风险后可以考虑入选–有室性心律失常病史或室性心律失常的风险因素,如结构性心脏病(例如,重度左心室收缩功能不全、左心室肥大)、冠心病(症状性或通过诊断检测证实缺血)、具有临床意义的电解质异常(例如,低钾血症、低镁血症、低钙血症)或有长QT综合征家族史。
6. 被诊断为IV期乳腺癌或炎性乳腺癌的受试者。
7. 有任何既往(同侧和/或对侧)浸润性乳腺癌或DCIS病史的受试者。有对侧DCIS病史且在任何时间仅接受过局部区域治疗的受试者可能有资格参加研究。
8. 受试者在筛选前5年内患有或有任何其他恶性肿瘤病史,经适当治疗的宫颈原位癌、非黑色素瘤皮肤癌或I期子宫癌除外。o对于在随机化日期前5年内有其他非乳腺癌病史且经研究者判断认为复发风险非常低(例如,接受手术治疗的甲状腺乳头状癌)的受试者,应与申办方讨论确定是否符合研究资格。
9. 既往接受过选择性ER下调剂或降解剂或AI内分泌治疗的受试者–如果受试者在进入研究时正在接受或模拟标准辅助内分泌治疗,可在末次非内分泌治疗(手术或化疗)后接受最多4周的内分泌治疗,直至随机化,以后发生者为准。
10. 存在已知符合Child-PughB级或C级标准的具有临床意义的肝脏病史,包括肝炎(例如,乙型肝炎病毒[HBV]或丙型肝炎病毒[HCV])、目前酗酒、肝硬化或病毒性肝炎检测呈阳性的受试者,具体定义如下:–活动性感染定义为需要抗病毒治疗或乙型肝炎(乙型肝炎表面抗原和/或总乙型肝炎核心抗体[HBcAb])或HCV抗体检测结果呈阳性。如果既往未进行过HBV或HCV评估,则受试者在筛选时无需进行这些评估,但当地法规要求的情况除外。–对于HBcAb检测呈阳性的受试者,仅在乙型肝炎表面抗体检测结果也呈阳性且HBVDNA聚合酶链反应呈阴性时,有资格参加研究–对于HCV血清学呈阳性的受试者,仅当HCVRNA检测结果呈阴性时,有资格参加研究。
11. 已知对任何研究药物或其任何辅料过敏或有超敏反应的受试者。
12. 已知对LHRH激动剂有超敏反应的绝经前和围绝经期受试者或男性受试者。
13. 已知存在口服药物吞咽问题的受试者。
14. 有出血体质、凝血障碍或血栓栓塞病史记录的受试者。
15. 受试者患有吸收不良综合征或其他可能干扰肠内吸收的疾病。
16. 受试者患有未控制的炎性肠病或慢性腹泻、短肠综合征或有重大上消化道手术史。
17. 受试者在随机化前28天内接受过与乳腺癌无关的重大手术,或预期将在研究治疗期间需要接受重大手术。
18. 受试者在筛选前14天内发生需要口服或静脉注射抗生素的严重感染,或在筛选前14天内发生其他具有临床意义的感染(例如,COVID-19)在筛选前至少14天,从严重或具有临床意义的感染中完全恢复的受试者有资格参加研究。如果受试者表现出潜在COVID-19感染的体征或症状,并有合理理由怀疑发生感染,研究者应遵循美国临床肿瘤学会2020年指南或机构指南进行检测。
19. 依据研究者的判断,受试者在临床实验室检查中存在妨碍其安全参与和完成研究的任何严重疾病或异常。
20. 研究者认为受试者不能或不愿意遵守方案要求。






 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡